

两种强启动子在枯草芽孢杆菌中调控表达研究
- 时间:2016-09-08
周艳敬 常晓娇 吴子丹 伍松陵 孙长坡
玉米赤霉烯酮(Zearaleonoe,ZEN)是由禾谷镰刀菌产生的一种非甾体霉菌毒素,广泛存在于受污染的小麦、玉米、高粱等谷物及其制品中。研究表明,ZEN具有生殖毒性、肝脏毒性、免疫毒性和遗传毒性,可导致动物生殖障碍、繁殖功能紊乱,人食用被其污染的食物会导致肝癌、食道癌和青春期早熟等疾病。传统的ZEN脱毒方法有高温、辐照、压煮等物理法及氨化、臭氧处理、碳酸钠浸泡等化学法。但这些方法效果不稳定,营养成份损失较大,易造成二次污染。利用微生物产生的胞内、胞外酶将真菌毒素降解成无毒或毒性较低的物质具有去毒效率高、不破坏营养等优点。
目前,ZEN的生物脱毒研究主要侧重在降解菌株的筛选和降解能力的分析方面,而对于降解酶的高效表达和酶制剂的应用研究非常少。2002年,El-Sharkawy等发现粉红螺旋聚孢霉(Clonostachys rosea)菌株可以分泌专化内酯酶,能够将ZEN完全转化为无毒物质,随后其降解基因在酿酒酵母中的表达取得了成功。国内关于ZEN降解酶基因研究报道较少,刘海燕等从粉红粘帚霉中克隆到玉米赤霉烯酮降解酶基因ZLHY6,该基因在毕赤酵母(GS115)中进行表达后能够高效降解ZEN。随后,郝小龙等对该降解酶的酶学性质进行了表征。
在生物体的遗传系统中,启动子起着重要的作用,选用强启动子介导目的基因的表达是提高基因异源表达非常有效的方法。P43启动子是来源于枯草芽孢杆菌的组成型强启动子,目前在以枯草芽孢杆菌为宿主菌的外源蛋白表达的研究和应用中使用广泛。杨明明等以β-半乳糖苷酶为报告基因,筛选得到能够在枯草芽孢杆菌中高效表达的启动子PlapS,其表达的酶活性比P43启动子有很大提高,具有良好的应用前景。
本实验室已经完成了ZEN的降解酶基因ZLHY6在多个表达系统中的高效表达,为了适应实际应用的需要,目前已将该基因克隆到载体pWB980中,得到重组表达载体pWBZ1,电转化枯草芽孢杆菌Bs 168得到降解酶基因ZLHY6阳性转化子Z1,酶活测定结果显示在启动子P43的调控下ZLHY6能够分泌表达。本文研究了两种组成型强启动子在枯草芽孢杆菌Bs 168 中介导的ZEN降解酶表达水平的差异,为ZEN降解酶基因在枯草芽孢杆菌中的高效异源表达奠定基础。
1 材料与方法
1.1 材料
1.1.1 实验材料
枯草芽孢杆菌Bs 168和表达载体pWB980为本实验室保存,其中pWB980为带有启动子P43及信号肽(SacB)片段的分泌表达载体。pWBZ1为含有ZLHY6降解酶基因的pWB980载体;启动子PlapS及信号肽SacB DNA片段由上海英骏生物技术有限公司合成,并由克隆载体pMD18-T Vector携带转化入大肠杆菌DH5α中。
1.1.2 培养基及生化试剂
本研究所用限制性内切酶、T4 DNA Ligase均购自TaKaRa公司;胶回收试剂盒和质粒提取试剂盒购自AXYGEN公司;乙腈、甲醇均为色谱纯,购自迪马公司;ZEN标准品购自Sigma公司;KOD-Plus及其它试剂,购自北京鼎国昌盛生物技术有限责任公司。
枯草芽孢杆菌的培养和发酵均使用LB培养基。LB培养基:胰蛋白胨10g、酵母提取物5g、NaCl 10g,去离子水定容至1L,121℃灭菌30min,室温保存备用;LB固体培养基:于液体培养基中加入1.3%~1.5%琼脂。枯草芽孢杆菌感受态细胞的制备使用BHI(Brain Heart Infusion)培养基,购自BD公司;50mg/mL卡那霉素(Kan):称取0.5g固体硫酸卡那霉素,用去离子水定容至10mL,0.2μm进口滤膜过滤除菌,分装后-20℃保存,使用终浓度为50μg/mL。
1.1.3 仪器设备
waters E2695型液相色谱仪(荧光检测器、C-18色谱柱250mm×4.6mm×5μm),waters公司;BIO C1000型PCR仪,BIO-RAD公司;EVOLUTION 300全波长分光光度计,Thermo公司。
1.2 实验方法
1.2.1 启动子及信号肽片段的克隆
根据PlapS启动子和信号肽的序列,利用Vector NTI软件设计合成一对引物,上游引物:F:5'-CCGGAATTCTCAGGAGCATTTAACCTAAA-3';下游引物:R:5'-GGCAAAAGCTTGAGTTGC-3',其中下划线分别为EcoR I和HindⅢ酶切位点,扩增片段为395bp。引物由北京华大基因股份有限公司合成。以携带PlapS和信号肽片段的DH5α为模板,PCR反应体系为:10×Buffer 5μL,MgSO4(25mmol/L)3μL,dNTP(2mmol/L)5μL,上、下游引物各1.5μL,KOD-Plus 1μL,模板DNA 1 μL,用无菌双蒸水补足至50μL。PCR反应条件为:94℃预变性5min;94℃1min,50℃30s,68℃30s,共31个循环;68℃延伸10min。1%的琼脂糖凝胶电泳检测,纯化PCR产物。
1.2.2 表达载体pWBZ7 的构建
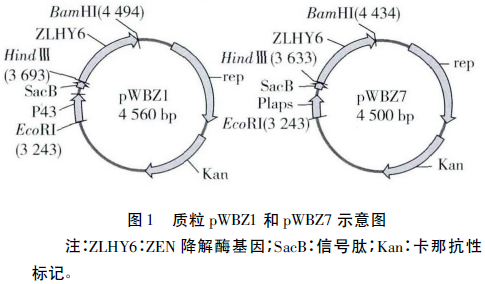
用质粒提取试剂盒提取菌株Z1的质粒pWBZ1,该质粒的P43启动子和信号肽序列位于EcoR I和Hind Ⅲ酶切位点之间,ZLHY6基因位于Hind Ⅲ酶切位点下游。pWBZ1经EcoR I和Hind Ⅲ 37℃过夜酶切。酶切体系为(30μL):质粒10μL,EcoRI 1.5μL,Hind Ⅲ 1.5μL,10×M Buffer 3μL,ddH2O 14μL。酶切产物经核酸电泳后切取大片段,用DNA胶回收试剂盒回收。
回收后的目的片段用限制性内切酶EcoR I 和Hind Ⅲ双酶切,酶切体系为(30μL):PCR产物2.5μL,EcoR I 1.5μL,Hind Ⅲ 1.5μL,10×M Buffer 3μL,ddH2O 21.5μL,37℃温浴过夜。回收纯化酶切产物,并检测纯化后的浓度。
回收后的两个片段经T4 DNA连接酶连接,连接反应体系为(10μL):pWBZ1片段5μL,PlapS启动子和信号肽片段3μL,10×T4 DNA连接缓冲液1μL,T4 DNA连接酶1μL,16℃连接10h。对酶连产物醇沉处理:在10μL酶连产物中加25μL预冷的无水乙醇,-20℃放置4h,于4℃ 12000r/min离心20min。缓慢弃去上清,DNA沉淀晾干后用6μL去离子水重悬连接产物。
1.2.3 工程菌Z7 的构建
1.2.3.1 枯草芽孢杆菌(Bs 168)感受态细胞的制备
挑取Bs 168单菌落,接种于5Ml LB试管培养基中,37℃,220r/min培养12h。按1%接种量转接至含50mL BHI培养基的三角摇瓶中,于37℃,220r/min培养,将菌液培养至OD600达到0.8时,约3h。将细菌培养物于4℃、7000r/min离心5min。用预冷的无菌水将菌体沉淀重悬,7000r/min离心5min,重复三次。用2mL无菌的40% PEG4000将菌体沉淀重悬。充分混匀后分装,每管150μL,-80℃冰箱保存备用。
1.2.3.2 连接产物的转化
取连接产物3μL,加入Bs 168感受态细胞150μL。电转化条件为:2.2kV,1000Ω,25mF,电击后,迅速向转化体系中加入800μL LB液体培养基,并转至1.5mL的离心管中,37℃复苏3h。将菌悬液涂布于Kan抗性LB平板,37℃培养,筛选阳性转化子。挑取转化子接种至LB液体培养基。37℃,220r/min培养过夜,分别对转化子进行PCR、重组质粒酶切鉴定及降解酶活性检测。重组质粒命名为pWBZ7,转化子命名为Z7。
1.2.4 ZEN 工程菌Z1、Z7的分泌表达
将新鲜平板上的Z1和Z7单菌落分别接入5mL Kan抗性LB液体培养基中,37℃培养12h,分别检测两菌株种子液的OD600,将Z1和Z7以相同接菌量转接于含有Kan抗性的50mL LB液体培养基中。分别于37℃、220r/min发酵培养2、4、6、8、10、12、15、23、30、35、47、54h,取发酵液,用于酶活测定和SDS-PAGE分析,将含空载体的Bs 168作为阴性对照,发酵过程中监测两菌株菌浓变化。
1.2.5 ZEN 降解酶活性测定
将ZEN标准品溶液分装于2mL的EP管中,含量为每管10μg,样品发酵液稀释2.5倍后取500μL与ZEN于37℃、220r/min共培养20min。参照GB/T 23504—2009[14],高效液相色谱检测ZEN含量。检测条件为:柱温箱25℃,荧光检测器激发波长274nm,发射波长440nm,流动相为乙腈—水(50∶50,V/V),流速为1mL/min,进样量10μL。根据ZEN的残留量计算工程菌对ZEN的降解率和酶活。酶活定义为:1mL发酵液在37℃、220r/min条件下,1h降解1μg ZEN所需的酶量为一个酶活单位。
1.2.6 发酵上清液的SDS-PAGE检测
取样品发酵液1.5mL,离心后吸取发酵上清液1.3mL于1.5mL离心管中,TCA法浓缩蛋白。将20μL蛋白浓缩产物与5μL的5×SDS-PAGE上样缓冲液混合均匀,沸水浴10min。样品浓缩30倍,上样量为10μL。
1.2.7 重组质粒的稳定性分析
重组质粒pWBZ7的稳定性分析参照文献进行。
1.2.8 启动子的结构分析
结合BPROM(Softberry)软件对启动子P43和PlapS进行结构分析。
2 结果与分析
2.1 表达载体pWBZ7的构建与重组子鉴定
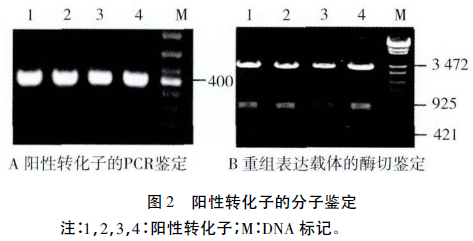
EcoR I 和Hind Ⅲ双酶切的启动子和信号肽片段与载体片段经连接(图1)、电转化入Bs 168感受态细胞中,涂布于卡那抗性平板,37℃培养12h。阴性对照无菌落长出,挑取目标平板阳性菌落,分别接LB液体培养基培养后进行PCR及质粒酶切鉴定。转化子经目的片段PCR扩增后在400bp处有清晰条带(图2 A) 。转化子质粒经EcoR I、BamH I 和Hind Ⅲ酶切后被切成大小分别为3310bp的载体片段、795bp的ZLHY6降解酶基因片段和大小为395bp的目的片段(图2B),与理论结果相符。测序分析与实验结果完全一致,证明PlapS和SacB片段已成功插入pWBZ1,载体pWBZ7及工程菌Z7构建成功。
2.2 重组子Z7酶活性验证
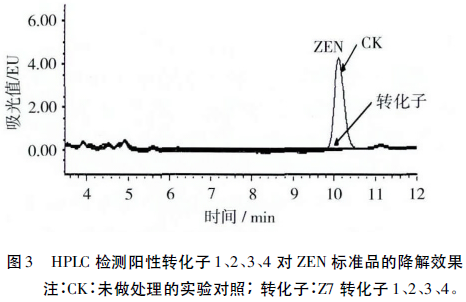
取转化子1、2、3、4的菌液各500μL分别加入ZEN毒素管,终浓度20μg/mL,对照中加入500μL LB。37℃、220r/min反应3h后,液相色谱检测ZEN残留量,检测结果见图3。由图3可看出,对照的ZEN出峰时间在10min后,样品中无ZEN检出,说明经几个转化子处理的ZEN毒素均降解完全,启动子PlapS能够调控ZEN降解酶基因在枯草芽孢杆菌中的表达。
2.3 ZEN工程菌的摇瓶发酵和降解酶的活性
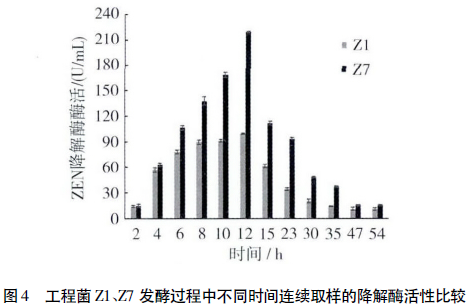
Z1、Z7发酵液经不同时间取样后立即进行ZEN标准品的降解实验。每个处理三次重复,检测结果取平均值。酶活测定结果显示,含空表达载体的Bs 168对ZEN无降解作用,工程菌Z1、Z7对ZEN具有明显的降解活性。两菌株的降解活性都在12h达到最高,此时Z7的ZEN降解酶的酶活是Z1的2.2倍,分别为219.02U/mL和99.6U/mL(图4),之后随着细胞活力衰退和目的蛋白被宿主菌分泌到胞外的蛋白酶分解,降解酶活性也随之降低。在发酵过程中,同时监测了不同时间发酵液菌体浓度的变化,发现两发酵液在发酵过程中菌体浓度基本保持相同,并在开始发酵10~35h过程中,Z7的降解酶活性较Z1均有明显提高,说明重组子Z7中的ZEN降解酶基因ZLHY6在枯草芽孢杆菌中启动子PlapS的介导下得到了加强表达。
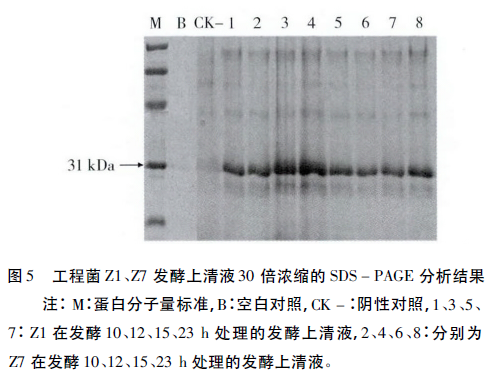
2.4 ZEN降解酶SDS-PAGE分析
将工程菌Z1、Z7发酵中期样品发酵液取上清浓缩后进行检测,SDS-PAGE分析结果显示,在目标位置有清晰条带(图5),与从氨基酸推断出的ZEN降解酶的理论分子质量大小一致。说明ZEN降解酶在启动子PlapS和P43调控下均得到了表达。
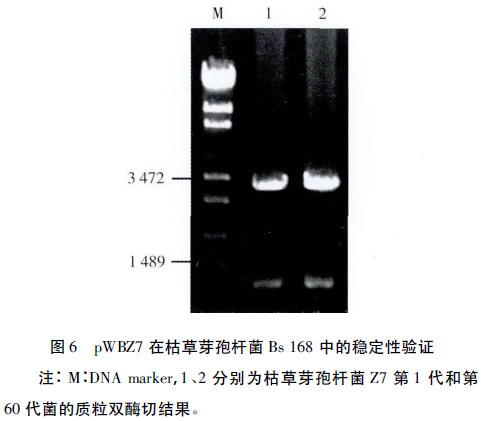
2.5 重组质粒的稳定性分析
将含有表达载体pWBZ7的枯草芽孢杆菌Bs 168转接于LB液体培养基,无抗性,37℃220r/min条件下培养,同时每24h转接一次,5d后将菌液(第60代菌)稀释106倍涂布LB平板,37℃培养,待菌落长出后随机挑选100个单菌落复制到Kan抗性LB平板,37℃培养。结果显示,工程菌Z7在Kan抗性LB平板上100%生长,提取第60代菌以及第一代菌的质粒进行双酶切验证,结果显示两者所含质粒相同(图6),说明重组质粒pWBZ7在枯草芽孢杆菌中非常稳定。
2.6 两种启动子的结构差异分析
PlapS启动子序列为246bp,由一个杂合启动子和一个Pluxs启动子构成,杂合启动子位于Pluxs上游,保守序列-10区TATTAT和-35区ATGATA分别来自于地衣芽孢杆菌PluxS启动子上游序列和枯草芽孢杆菌Papr启动子,保守区之间的间隔为17bp。Pluxs启动子结构为-35区TGAAAA和-10区TATAAT,保守区之间的间隔为17bp,被σA(σ55)因子识别;P43启动子序列为312bp,含双启动子结构,被两种RNA聚合酶的σ因子所识别,有被σB(σ37)识别的-35区AGAAAT和-10区GCGATT,间隔15bp,还有被σA(σ55)识别的-35区GTGAAA和-10区TAAAAT,间隔17bp。对PlapS和P43的启动子区的保守性进行分析发现P43被σA识别的启动子区和Pluxs启动子区的保守率相同(-35区66.7%,-10区83.3%)。SacB序列位于P43与PlapS下游,富含AG的SD序列位于SacB上游10bp处(图7)。

3 结论
启动子的结构是影响其表达活性的关键因素。一般来说,启动子和保守区之间的间隔为17bp时该启动子的活性最强,偏离17bp时启动子活性会变弱。此外,启动子保守序列与σ因子识别保守序列之间的相似程度越高,其表达能力也就越强。P43被σA识别的启动子区和PlapS的Pluxs启动子区间隔相同,保守率也相同。不同的是PlapS的杂合启动子和P43被σB识别的启动子区,这两个启动子保守区间隔分别为17bp和15bp,这可能是PlapS表达活性高于P43的原因之一。
实验中构建的质粒pWBZ7是将表达载体pWBZ1降解酶基因ZLHY6上游的启动子P43替换为PlapS,质粒其余部分完全相同,两个表达载体均转化枯草芽胞杆菌Bs 168进行表达。由于这两株工程菌发酵及取样处理ZEN毒素的方法和条件均相同,两个启动子均为组成型启动子,因此两种质粒介导的降解酶基因表达水平的不同取决于两个启动子的强弱。实验结果表明降解酶基因ZLHY6在两个启动子调控下都得到了高效表达,整个发酵期间的12次取样酶活测定结果显示PlapS的表达活性均高于P43,12h时酶活最高,PlapS为P43的两倍。而且重组质粒pWBZ7在枯草芽孢杆菌Bs 168中非常稳定。因此,较P43启动子,PlapS是一个活性更强的启动子,为介导枯草芽孢杆菌表达系统中异源基因的高效表达奠定基础。
本研究通过构建含PlapS强启动子的ZEN降解酶表达载体,获得一株ZEN降解酶高效表达工程菌,对不同强启动子调控降解酶基因表达的研究和真菌毒素的降解有非常重要的意义。
| 相关附件 |
